In mice, gene editing repairs a mutation that causes rare liver disorder
Article | April 14, 2026
The same approach helped treat Baby KJ Muldoon in 2025
(Bar Harbor, Maine – April 14, 2026) – Scientists have corrected an extremely rare and life-threatening genetic disease of the liver in mouse models and human patient cells, using the gene-editing approach that served as the basis for the historic, life-saving treatment of Baby KJ Muldoon in 2025.
Led by scientists from the Rare Disease Translational Center (RDTC) at The Jackson Laboratory (JAX), the Broad Institute, and the University of Southern California, the research lays the foundation for a potential new therapy for Zellweger spectrum disorder (ZSD), an incurable disease that affects 1 in 50,000 to 90,000 births in North America.
The findings appear today in Nature Biomedical Engineering.
“This is about more than correcting a single mutation,” said RDTC Vice President Cathleen (Cat) Lutz, a co-senior author of the study. “It’s about building a new paradigm for how we develop genetic therapies, one that starts with precise disease models, scales through platform technologies like base editing, and ultimately reaches patients faster.”
A genetic disorder with deep implications for the liver and central nervous system, ZSD is caused by mutations in PEX genes that make cell structures called peroxisomes, which break down fats and remove toxins. When peroxisomes don’t function properly, the liver, brain, and other organs can become damaged.
The team showed that base editing, which corrects individual DNA letters without cutting DNA, can repair mutations in the PEX1 gene that cause ZSD. The study demonstrated that this edit restored function of the liver and peroxisomes when made in mice and patient-derived cells modeling the disorder.
The scientists also showed that the efficiency of the base editors increased over the duration of the study. A lower dose of base editors was able to achieve the same level of editing over time as the higher dose tested by the team.
“We are really excited about the possibility of using this tool that can correct a genetic error. If we can do that in patients, doctors won’t only treat symptoms like they do today. Moving these therapeutics to the clinic means we will be fixing the underlying problem with a permanent treatment,” said Maximiliano Presa, a lead scientist at JAX’s RDTC who co-led the work.
Advancing therapeutic technologies
The breakthrough builds on a long-standing collaboration led by Cat Lutz alongside David Liu, a core member of the Broad Institute; Joseph G. Hacia, a medical geneticist from the Keck School of Medicine of the University of Southern California; Nancy Braverman, a clinician scientist at McGill University; and Ann Moser, a research associate in neurology at Kennedy Krieger Institute and Johns Hopkins University.
“I am honored to collaborate with the outstanding teams led by David Liu at the Broad Institute and Cat Lutz at The Jackson Laboratory on this work,” said Hacia, a co-senior study author. “There is a critical unmet need for therapies that address the root causes of genetic disease. Our findings in a common form of ZSD point to a potential path forward that may extend to other conditions, and we look forward to continued efforts to advance this approach toward clinical translation. Liver disease remains a major driver of morbidity and shortened lifespan in ZSD, and by targeting the underlying genetic defect, we aim to deliver meaningful, lasting improvements in patient health.”
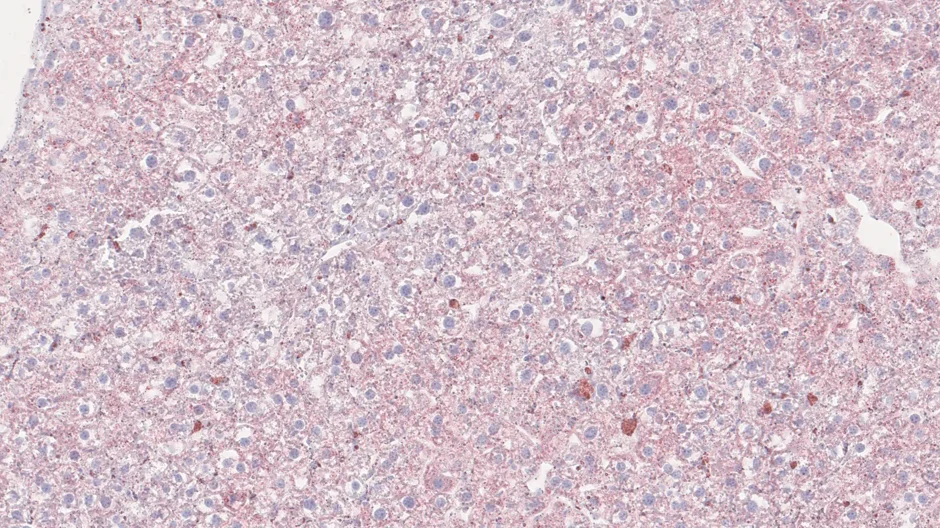
Microscopy images of mouse liver tissue with and without Zellweger spectrum disorder (ZSD). Tissue from mice with ZSD shows an increased accumulation of fats, or lipids, which correspond to redder hues. When the mice were treated by editing the PEX1 gene, liver tissue returned to near-normal levels.
The team focused on improving mouse models that mirror how ZSD develops in humans. Earlier models provided critical insights into disease biology, although their limited survival and variability made it challenging to study longer-term disease progression in a consistent way. In contrast, the newly developed models from the Lutz lab survive and develop liver dysfunction that closely mirrors the disease, including its very long chain fatty acid biomarkers. The combination of treatments and a longitudinal biomarker increases the team’s confidence that the results could translate to patient care.
The researchers tested two different versions of the base editors in mice using viruses that delivered them directly to the liver. Both editors efficiently corrected the Zellweger mutation, but one called ABE8e-V106W was significantly better tolerated in the animals and made fewer unwanted edits.
By applying the editors to neonatal and older mice with the mutation, the team observed correction of the PEX1 gene in roughly 60% of liver cells. This was high enough to restore peroxisome and liver function and lower toxic buildup of potentially toxic peroxisomal metabolites in circulation, liver, and/or brain. In cells derived from human patients, the editor corrected more than 80% of PEX1 mutations and restored peroxisome homeostasis. The team also found evidence that correcting the PEX1 gene deficiency in the liver reduced the buildup of potentially toxic branched-chain fatty acids in the brain, an early observation that suggests treating the liver could also address neurological aspects of disease.
“What we observed in mouse livers with this permanent mutation fix was especially encouraging,” Presa said. “Next, we will explore different delivery modalities that in addition to the liver, will provide broader accessibility to the central nervous system. From a patient’s perspective, this has the potential for multiple organ system benefit that could impact quality of life issues, such as improved hearing and eyesight.”
In 2025, before the study was published, the early observations were compelling enough that the researchers recommended using the same gene-editing technology in Baby KJ’s treatment. The approach corrected a different mutation, helping save the newborn from a life-threatening rare metabolic disorder.
Lutz’s team plans to move the research towards more comprehensive treatment options. The idea is to develop and validate genetic therapies that can effectively treat severe genetic diseases like ZSD, move these into the latest FDA framework, and scale the application to treat more rare diseases more quickly.
“We’ve spent nearly a decade building deep expertise in rare diseases, not just by making patient mouse and cell models using them to demonstrate the safety and efficacy of emerging therapeutics, this is work that ultimately reshapes how clinical trials are designed and patients are treated today,” Lutz said. “We are looking forward to the next chapter with our collaborators at the Broad—moving these therapeutics into the clinic.”
This work was supported by NIH Somatic Cell Genome Editing (SCGE) Collaboration Opportunity Fund (COF), NIH grants U01 AI142756, RM1 HG009490, and R35 GM118062, and HHMI; NIH SCGE COF, the Center for Precision Genetics at The Jackson Laboratory (NIH grant U54 OD020351 and U54 OD030187) and the Mouse Mutant Resource and Research Center (NIH grant U42OD010921); NIH grant R24 OD030033, the Global Foundation for Peroxisomal Disorders and Wynne Mateffy Research Foundation.
We acknowledge that the earlier PEX1-mutant mouse model, which laid the foundation for the Lutz laboratory’s work, was supported by Anne Park and Matt Hopkins through their Pound the Pavement for Peter fundraiser in honor of their son Peter, as well as Denise and Woody Woodbury.
Other authors are: Xin D. Gao, Maximiliano Presa, Jordyn E. Duby, Jennifer Ryan, Pierre-Alexandre Piec, Alvin Hsu, Samagya Banskota, Allen Yujie Jiang, Lingxiao Chen, Gregory A. Newby, Erminia Di Pietro, Jonathan M. Levy, Bradford H. Steele, Sarah Lecordier, Fangfei Qin, Ann B. Moser, Jun Xie, Guangping Gao, Nancy E. Braverman, Aamir R. Zuberi, and David R. Liu.
JAX media contact: Roberto Molar, [email protected], 202-765-5144
About The Jackson Laboratory
The Jackson Laboratory (JAX) is an independent, nonprofit biomedical research institution with a National Cancer Institute-designated Cancer Center. JAX leverages a unique combination of research, education, and resources to achieve its bold mission: to discover precise genomic solutions for disease and empower the global biomedical community in the shared quest to improve human health. Established in Bar Harbor, Maine in 1929, JAX is a global organization with nearly 3,000 employees worldwide and campuses and facilities in Maine, Connecticut, California, Florida, New York, and Japan. For more information, please visit www.jax.org.
Citation: Gao XD, Presa M, et al. In vivo base editing rescues liver pathophysiology and peroxisome dysfunction in a mouse model of Zellweger spectrum disorder. Nature Biomedical Engineering. (2026). https://www.nature.com/articles/s41551-026-01651-5
Learn More
Genome editing corrected rare brain mutations in mice. Could it help fight neurological diseases?
Scientists have successfully corrected gene mutations in mice with an ultra-rare disease by editing DNA directly in the brain using a single injection. This technique, which also showed promise in patient-derived cells, addressed mutations causing alternating hemiplegia in childhood (AHC), reducing symptoms and extending survival in affected mice.
View more
What rare diseases can teach us about the rest of medicine
Why genetic outliers might hold the key to common conditions—and why we should invest more in understanding them
View more